


La vaccinovigilanza, come è noto, si basa sulle informazioni raccolte tramite il sistema di farmacovigilanza passiva, dove confluiscono le segnalazioni spontanee delle sospette reazioni avverse dopo vaccinazione (AEFI; Adverse Event Following Immunization1), e tramite il sistema di farmacovigilanza attiva, laddove vengano predisposti specifici studi post-marketing che indaghino attivamente gli eventi post vaccinazione per un determinato lasso temporale.
La presenza di questi progetti può portare ad un aumento significativo del numero di segnalazioni raccolte dalla Rete Nazionale di Farmacovigilanza ma non è noto se siano stati definiti appositi progetti di farmacovigilanza attiva sulla profilassi anti COVID-19, nonostante la mancanza di dati a supporto e consolidamento delle evidenze di efficacia e sicurezza di questi farmaci.
Ciò pone anche importanti interrogativi riguardo ai numerosi casi di infezione e decessi riportati dalle cronache giornalistiche a breve distanza dalla prima o seconda dose del trattamento (vedi “Bollettino basato su eventi in Italia”): questi casi, certamente, sarebbero oggetto di grande attenzione nell'ambito di studi di farmacovigilanza attiva, mentre non vi è alcuna garanzia che siano adeguatamente indagati nel caso in cui la sorveglianza si basi su segnalazioni spontanee.
Laddove è iniziata la campagna di vaccinazione del personale scolastico, inoltre, si registrano punte del 90% di assenze dal lavoro (vedi “Bollettino basato su eventi in Italia”) per gli effetti collaterali subiti a seguito della somministrazione del farmaco.
La domanda, quindi, sorge spontanea: tutti questi eventi stanno confluendo nella Rete Nazionale di Farmacovigilanza?
Emblematico è il caso fatale della dott.ssa Gabriela Godoy2, farmacista e madre di quattro figli, sentitasi male il giorno dopo aver effettuato il Comirnaty e morta dopo 5 giorni di ospedalizzazione. Si apprende che si è trattato di un ictus: non è stato effettuato alcun esame autoptico, piuttosto i suoi organi sono stati espiantati per la donazione.
Può essere solo una coincidenza temporale la somministrazione di Comirnaty, un trattamento OGM sviluppato in pochi mesi con tecnologia mRNA che fino a oggi non era mai stata oggetto di licenza da parte delle autorità regolatorie e pertanto mai testata su vasta scala?
EudraVigilance, la banca dati europea delle segnalazioni di sospette reazioni avverse3, offre un accesso pubblico per la consultazione di questi eventi collegati ai nuovi trattamenti di profilassi autorizzati in maniera CONDIZIONATA4 (CMA, Conditional Marketing Approval):
- Comirnaty (nome nella sperimentazione “BNT162b2”; nome generico “Tozinameran”) di Pzfizer- BioNTech, autorizzato dalla Commissione Europea il 21 dicembre 2020;
- Moderna COVID-19 Vaccine (nome nella sperimentazione “mRNA-1273”; nome generico “CX-024414”. Altri nomi: “COVID-19 Vaccine Moderna”, “COVID-19 mRNA Vaccine Moderna”) di Moderna, autorizzato dalla Commissione Europea il 6 gennaio 2021;
- COVID-19 Vaccine AstraZeneca (nome nella sperimentazione “ChAdOx1 nCoV 19 vaccine AZD1222”; nome generico “CHADOX1 NCOV 19”) di Oxford University e AstraZeneca, autorizzato dalla Commissione Europea il 29 gennaio 2021.
Al 5 marzo la banca dati EudraVigilance registra i seguenti casi individuali di sospette reazioni avverse segnalati nei Paesi dello Spazio Economico Europeo (in inglese European Economic Area, EEA):
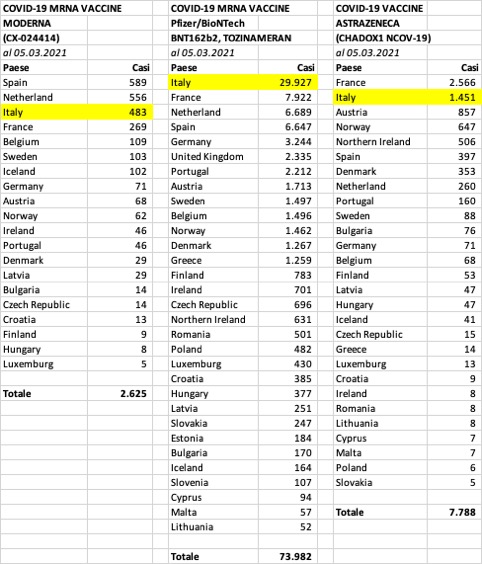
In Italia nell’arco di poco più di due mesi sono stati segnalati in totale 31.861 casi di sospette reazioni avverse.
Complessivamente (paesi EEA e non EEA) la totalità delle segnalazioni al 5 marzo sono:
- Pzfizer/BioNTech, Comirnaty, BNT162b2, Tozinameran: 92.859 casi;
- Moderna CX-024414: 4.552 casi;
- AstraZeneca, ChAdOx1 nCoV 19, AZD1222: 21.638 casi.
I dati riferiti al farmaco Comirnaty sono statisticamente rilevanti: al 5 marzo, secondo le elaborazioni fornite sui repository open data consultabili sulla piattaforma GitHub5, in Italia sono state somministrate 4.399.067 dosi del farmaco. Rapportando i dati dei casi individuali con le dosi somministrate si evidenzia un'incidenza di 680 casi di sospetta reazione avversa ogni 100.000 dosi somministrate.
Considerando che nel periodo preso in esame sono state somministrate in media 63.775 dosi di Comirnaty al giorno, si ricava che in media si effettuano 434 segnalazioni spontanee (casi individuali) di reazione avversa al giorno.
Nonostante i dati siano certamente sottostimati, in quanto riflettono le sole segnalazioni spontanee (cfr. farmacovigilanza passiva), si tratta di un'incidenza altissima e mai riscontrata sino a oggi per qualsiasi altro farmaco in commercio.
Dai dati pubblici presentati da EudraVigilance, purtroppo non è possibile stabilire quante di queste segnalazioni provenienti dalla nostra Rete Nazionale di Farmacovigilanza siano riferibili a casi gravi (serious) e non gravi (non serious): i dati che descrivono e analizzano le AEFI si presentano in forma aggregata e l’unica distinzione è rappresentata dalla provenienza geografica distinta tra Paesi dello Spazio Economico Europeo e Paesi al di fuori dello stesso (quali siano questi Paesi e perché questi dati confluiscano nella banca dati europea non è dato sapere). Alcuni dati che si possono desumere da queste segnalazioni lasciano ulteriormente perplessi:
- L’Italia è il Paese che contribuisce maggiormente e in misura notevolissima alle segnalazioni spontanee rispetto agli altri Paesi dello Spazio Economico Europeo, con una quota complessiva del 37,8% (il 40,5% per il vaccino Pfizer/BioNTech, il 18,4% per il vaccino Moderna e il 18,6% per il vaccino Astra Zeneca);
- La stessa incidenza delle segnalazioni è anomala, in particolare riguardo al vaccino Pfizer/BioNTech: rispetto alla Germania ad esempio (al 5° posto in questa particolare classifica delle segnalazioni spontanee) l’Italia ha una incidenza, come detto sopra, di 680 segnalazioni ogni 100.000 dosi somministrate, contro appena 52 della Germania (oltre 10 volte);
- Sulla totalità delle segnalazioni raccolte da Eudravigilance relative alla somministrazione del vaccino Pfizer/BioNTech (92.859), circa l’83% appartengono ai paesi EEA: se guardiamo invece alla somministrazione del vaccino Moderna, questa proporzione cambia (EEA=59%) e addirittura si rovescia per il vaccino Astra Zeneca (EEA=37%);
- Spicca il numero delle reazioni avverse del vaccino AstraZeneca rispetto al vaccino Moderna che sono stati distribuiti nei Paesi EEA quasi contemporaneamente (le segnalazioni sono in un rapporto superiore a 4:1)
Per maggiori dettagli sulle segnalazioni di reazione avversa dei trattamenti di profilassi anti covid-19 in Italia e relative analisi dei dati occorre attendere i report mensili dell’Agenzia Italiana del Farmaco (AIFA). Il primo rapporto pubblicato lo scorso 4 febbraio6 (riferito al periodo 27 dicembre – 26 gennaio) si presenta purtroppo piuttosto scarno e sembra più mirato a rassicurare piuttosto che a informare: consultando i dati si nota l’assenza di un disegno di studio a monte, non si ha evidenza del numero dei casi per tipologia di reazione e per fascia di età (a titolo di esempio, nessun dato è riportato sulle gravi reazioni allergiche, sulle positività a Sars Cov-2 riscontrate dopo la somministrazione, … ), il tasso di segnalazione è rapportato soltanto al numero di dosi somministrate e non anche agli individui vaccinati (a cui vengono somministrate due dosi), si indica che il 4% delle segnalazioni proviene da progetti di farmacovigilanza attiva ma non si dettagliano né tantomeno si confrontano i tassi di segnalazione con quelli provenienti dalle segnalazioni spontanee.
Alla data odierna, 8 marzo 2021, non è ancora disponibile il secondo rapporto mentre nel Regno Unito è già pubblicato il quinto aggiornamento7 e non sappiamo se i dati verranno presentati anche in forma cumulativa (gennaio + febbraio). Una cosa è certa: sarà impossibile confrontarli con i dati raccolti dalla banca dati EudraVigilance, sia perché i periodi di riferimento non potranno coincidere, sia perché come già menzionato i dati della banca dati europea vengono presentati in forma aggregata.
Nella tabella che segue si riportano i dati aggregati (dal 27 dicembre al 5 marzo) riferiti ai tre trattamenti di profilassi approvati dalla Commissione Europea con licenza CMA suddivisi in base alla gravità. Si riporta, inoltre, il dato riferito ai casi con esito fatale (decesso).
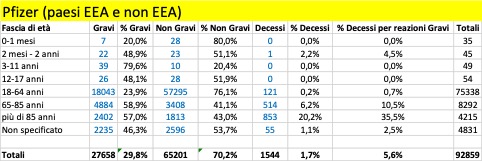
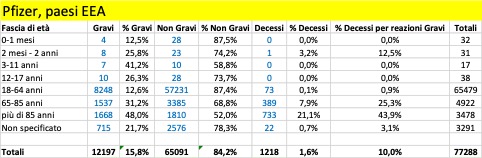

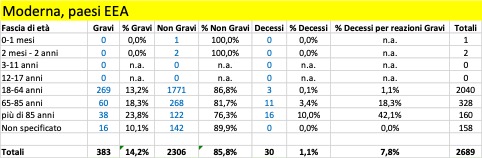
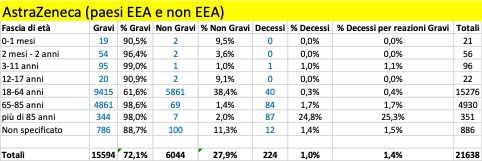
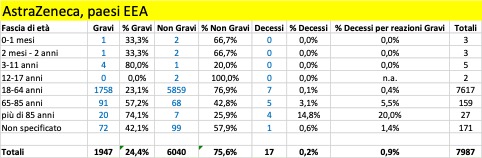
Come si può osservare, per tutti e tre i trattamenti i casi classificati gravi che confluiscono nella banca dati europea si verificano per la maggior parte nei Paesi al di fuori dello Spazio Economico Europeo (Pfizer 12.197 EEA su 27.658, Moderna 383 EEA su 2.241, Astrazeneca 1.947 EEA su 15.594) al contrario dei casi classificati non gravi che vengono riportati per la maggior parte nei Paesi EEA (per i farmaci Moderna e Astrazeneca rappresentano la quasi totalità dei casi classificati non gravi).
Il fenomeno, almeno in apparenza, si presenta piuttosto anomalo nel suo complesso.
Dai dati si evince che siano già in corso sperimentazioni sui bambini a cominciare dalla fascia di età 0 mesi – 1 mese e che si stiano registrando anche esiti fatali: un caso attribuito al trattamento Pfizer verificatosi nella fascia di età 2 mesi - 2 anni, un caso attribuito al trattamento Astrazeneca e un altro caso a quello di Moderna, entrambi avvenuti nella fascia di età 3 - 11 anni.
Per quanto riguarda gli esiti fatali si registrano in totale 2.164 decessi, di cui 1.265 provenienti da paesi EEA e 1.218 attribuiti al farmaco della Pfizer.
Complessivamente, dal 20% al 35% delle reazioni avverse portano alla morte delle persone con più di 85 anni: il più pericoloso in tal senso sembra essere il vaccino di Moderna. Se facciamo riferimento alle reazioni avverse considerate gravi, la forbice sale dal 25% a oltre il 50%: un anziano su due con più di 85 anni, vaccinato con il vaccino Moderna e che ha una reazione avversa grave, muore! Non sono messi molto meglio neanche le persone nella fascia 65-85 anni: qui, in caso di reazione avversa grave con i vaccini Pfizer/BionTech e Moderna la forbice della mortalità va dal 10,5% al 25,5%, mentre risulta più contenuta per i vaccinati con AstraZeneca, 1,7%. Ma in questo caso la ragione è certamente legata al fatto che il vaccino, finora, non è stato somministrato nella maggior parte dei paesi agli over 65: cosa succederà se verrà confermata la possibilità di somministrarlo anche a questa fascia di popolazione?
La fascia di età in cui si riscontra il maggior numero dei casi di reazione avversa è quella 18 – 64 anni, che presumibilmente rappresenta la fascia che ha ricevuto il maggior numero delle somministrazioni.
Come è noto, nell’ambito delle attività di sorveglianza, le segnalazioni di AEFI sono uno strumento fondamentale per il monitoraggio continuo della sicurezza dei farmaci.
A maggior ragione assume un ruolo di fondamentale importanza nel contesto di farmaci che hanno appena ottenuto un’approvazione condizionata, sulla base di dati di sicurezza osservati per pochi mesi (e non anni) emersi dai trial clinici pre-autorizzativi riferiti a un campione non rappresentativo del mondo reale8. La relazione finale dello studio cardine di fase 3, soggetta a Obbligo Specifico nel contesto della Licenza, infatti, potrà essere presentata dal titolare non prima di due anni dal CMA (Conditional Marketing Approval). A ciò va aggiunto che la fuoriuscita dei partecipanti dal trial di fase 39 inficerebbe la possibilità di completare/integrare gli studi e di confermare i benefici del farmaco in termini di sicurezza, tollerabilità, immunogenicità ed efficacia.
Un simile contesto richiede trasparenza (anche relativamente alla valutazione dei dati raccolti), tempestività e completezza di dati, disponibilità di open data.
Sebbene lo scorso 14 dicembre 2020 l’Agenzia Italiana del Farmaco in accordo con il Ministero della Salute e il Commissario Straordinario per l’emergenza Covid-19 abbia costituito il Comitato Scientifico per la sorveglianza post-marketing dei Vaccini Covid19 (CSV-Covid19)10, nominando
- 15 esperti indipendenti con numerosi compiti fra i quali
“esaminare obiettivi e metodi impiegati nei progetti di farmacovigilanza (ordinari e straordinari) sostenuti dall’AIFA al fine di assicurarne la coerenza e la qualità dei risultati, svolgendo anche una funzione di indirizzo e di monitoraggio per identificare eventuali criticità e suggerire soluzioni adeguate; formulare raccomandazioni all’AIFA, in base alle evidenze disponibili, sugli orientamenti strategici utili a garantire la sicurezza e a perseguire l’efficacia delle attività di vaccinazione nel Paese; a partire dalle attività di studio e monitoraggio, indicare la necessità di specifici approfondimenti e formulare proposte per la realizzazione di studi integrativi o per il riorientamento degli studi in corso”
- 6 osservatori designati dalle varie istituzioni nazionali e regionali coinvolte nella pianificazione e nella gestione della campagna di vaccinazione con il compito di garantire che le informazioni siano tempestive e complete
oggi, come suddetto, non è ancora disponibile il Rapporto sulla Sorveglianza dei trattamenti di profilassi anti COVID-19 del II mese di somministrazione, non è dato sapere alcunché sugli studi di farmacovigilanza attiva approntati né sui lavori del Comitato Scientifico per la sorveglianza post-marketing dei Vaccini Covid19.
Eppure, non è noto se questi farmaci proteggano il ricevente dall’infezione (“non si esclude la possibilità di essere infettati o reinfettati”11) o se addirittura in particolari circostanze possano potenziarne la malattia, non è noto se siano in grado di impedire la circolazione dell’infezione (“non si esclude la possibilità di essere veicolo di contagio”11)), non è noto se siano in grado di “proteggere” il ricevente dalle varianti del virus in circolazione, non è noto se possano favorire lo sviluppo di nuove varianti.
Gli unici dati disponibili nel nostro Paese, quasi in tempo reale (e in formato open), sono quelli riferiti alle consegne dei farmaci e alle dosi somministrate.
NOTE
1 In accordo con il Council for International Organisations of Medical Sciences (CIOMS), un AEFI è definito, come “un evento avverso (grave o non grave) di particolare interesse medico e scientifico specifico per un prodotto per il quale potrebbe essere appropriato un monitoraggio continuo e una comunicazione rapida da parte dello sperimentatore allo sponsor. (...) A seconda della natura dell’evento, deve essere garantita una comunicazione rapida alle Autorità regolatorie”
2 https://www.corriereromagna.it/ravenna-malore-improvviso-muore-farmacista-madre-di-4-figli/
4 La procedura di approvazione condizionata prevede che il titolare dell'autorizzazione potrà presentare dati aggiuntivi anche dopo l'autorizzazione all'immissione in commercio, contrariamente ad una normale autorizzazione all'immissione in commercio in cui tutti i dati sono presentati prima del rilascio dell'autorizzazione.
Il CMA (Conditional Marketing Approval) è valido per un anno, su base rinnovabile.
L’Autorizzazione è subordinata alla presentazione, entro un periodo di tempo definito e a determinate scadenze, di ulteriori dati a supporto e consolidamento delle evidenze di efficacia e sicurezza (Specific Obligations - SOs), derivanti sia da studi ancora in corso al momento dell’autorizzazione per acquisire i risultati finali anche su nuovi endpoint, sia dando avvio a nuovi studi.
5 https://github.com/owid/covid-19-data/blob/master/public/data/vaccinations/vaccinations.csv
8 La sperimentazione clinica di fase 3 del farmaco BNT162b2 (Comirnaty) è iniziata il 27 luglio 2020. Il farmaco è stato testato su 43.500 persone in sei Paesi. I risultati dello studio sono stati pubblicati su The New England Journal of Medicine con l'articolo Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine, da cui si evince che il periodo di osservazione degli eventi avversi è di circa 14 settimane dopo la seconda dose. Vedi https://www.nejm.org/doi/full/10.1056/NEJMoa2034577
9 Direttamente dalla voce di una volontaria allo studio, intervistata dalla redazione della trasmissione televisiva Report andata in onda lo scorso 25 gennaio su RAI 3, si è appreso che al gruppo placebo è offerta la somministrazione di Comirnaty:
“Ora che lo studio è completo io voglio il vaccino, mi faranno altri esami e se ero nel gruppo placebo mi daranno subito il vaccino perché i volontari possono saltare la coda”.
11 Vedi allegato "Documento ASL ROMA 19 febbraio"










